Case Report
Small cell carcinoma of the ovary with hypercalcemia: Case report and review of the literature

Gerday A1*, Marbaix E2, Squifflet JL1, Mazzeo F3 and Luyckx M1
1MD, Gynaecology, Clinique Universitaire saint Luc, Bruxelles, Belgium2MD, PhD, Anatomopathology, Clinique Universitaire saint Luc, Bruxelles, Belgium
3MD, Oncology, Clinique Universitaire saint Luc, Bruxelles, Belgium
*Address for Correspondence: Gerday A, MD, Gynaecology, Clinique Universitaire saint Luc, Bruxelles, Belgium, Email: [email protected]
Dates: Submitted: 26 June 2018; Approved: 02 July 2018; Published: 03 July 2018
How to cite this article: Gerday A, Marbaix E, Squifflet JL, Mazzeo F, Luyckx M. Small cell carcinoma of the ovary with hypercalcemia: Case report and review of the literature. Clin J Obstet Gynecol. 2018; 1: 035-044. DOI: 10.29328/journal.cjog.1001006
Copyright License: © 2018 Gerday A, et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.

Abstract
We describe here the case of a 23-year old woman with small cell carcinoma of the ovary of the hypercalcemic type (SCCOHT) with SMARC-A4 mutation who benefited from surgery in two steps leading to a total hysterectomy with bilateral salpingo-oophorectomy, omentectomy, pelvic and lombo-aortic lymph nodes dissection. She also received 6 courses of poly-chemotherapy after the surgery. A close follow-up was then performed by clinical examination every three months with determination of serum calcium and CA125 level as well as imaging with thoraco-abdominal CT scan. To date, the patient has a disease-free survival of more than 9 years. We also reviewed the literature on this topic and discuted the new diagnostic and prognostic genetic tool SMARC-A4 mutation.
Introduction
Small cell carcinoma of the ovary with hypercalcemia (SCCOHT) is a rare neoplasm of the ovary with an aggressive character resulting in a very poor prognosis. This tumour type mainly affects young patients, especially in the second and third decade of life [1]. Hypercalcemia is found in more than 60% of the cases, but is rarely symptomatic at the time of diagnosis. If hypercalcemia is present at the moment of diagnosis, it may serve as “tumour marker” for the follow-up of the patient [1,2]. Historically, Scully reported a first case of small cell carcinoma of the ovary with hypercalcemia in 1979 [3], but it is only in 1982 that Richard Dickersin and colleagues defined this ovarian tumor as a particular entity with 11 reported cases [2]. Later, a larger retrospective study by Robert H. Young et al., described 150 patients presenting a small cell carcinoma with hypercalcemia. To date, it is largest series reported in the literature [1]. More recently, mutation of the SMARC-A4 gene coding for Brg-1 protein was reported as characteristic of SCCOHT [4].
The treatment of this disease is primarily surgical, frequently associated to chemotherapy. In some cases, radiation therapy is given to patients. The prognosis of these patients is very poor with a 5-year survival rate of 10 % [1,5]. We report a patient successfully treated by surgery and combined chemotherapy, who remains disease-free more than six years after initial diagnosis.
Case Report
A 23-year old female patient consulted in our department for an abdominal pain since three weeks. She had one pregnancy with vaginal delivery a few months earlier and was subsequently under oral contraception. The clinical examination revealed a large pelvic mass, with abdominal pain and fever over 39°C. Transvaginal echography showed a heterogeneous mass of about 14 cm in diameter. An abdominal computed tomography (CT)-scan confirmed the heterogeneous mass and a small amount of liquid in the pouch of Douglas and parieto-colic gutters. There was no evidence of carcinomatosis (Figure 1). Chest CT-scan was unremarkable. Biological investigations revealed a serum calcium level at 13.57 mg/dl (normal range 8.80 - 10.40 mg/dl) with a parathyroid hormone at less than 3 pg/ml (normal: 16-81pg/ml). CA 125 level was slightly increased to 40.8 U/ml (normal<25U/ml), whereas carcino-embryonic antigen (CEA) and alpha-foetoprotein were normal. Because of a persistent significant abdominal pain, the large adnexal mass, fever and an altered general status, we decided to carry out a diagnostic laparoscopy without delay. At laparoscopy, the large heterogeneous mass of the right adnexa was twisted by one turn on its pedicle. The right fallopian tube was normal as well as the left adnexa and the uterus. Careful examination of the peritoneum showed no sign of carcinomatosis and the liver was normal (Figure 2). Given the suspicious nature of the ovarian lesion, conversion into laparotomy by Pfannestiel incision was decided and a right salpingo-oophorectomy was performed. Frozen section of the mass revealed a poorly differentiated aggressive anaplastic carcinoma. The procedure was accordingly completed by a right ilio-obturator lymphadenectomy associated with an infra-colic omentectomy.
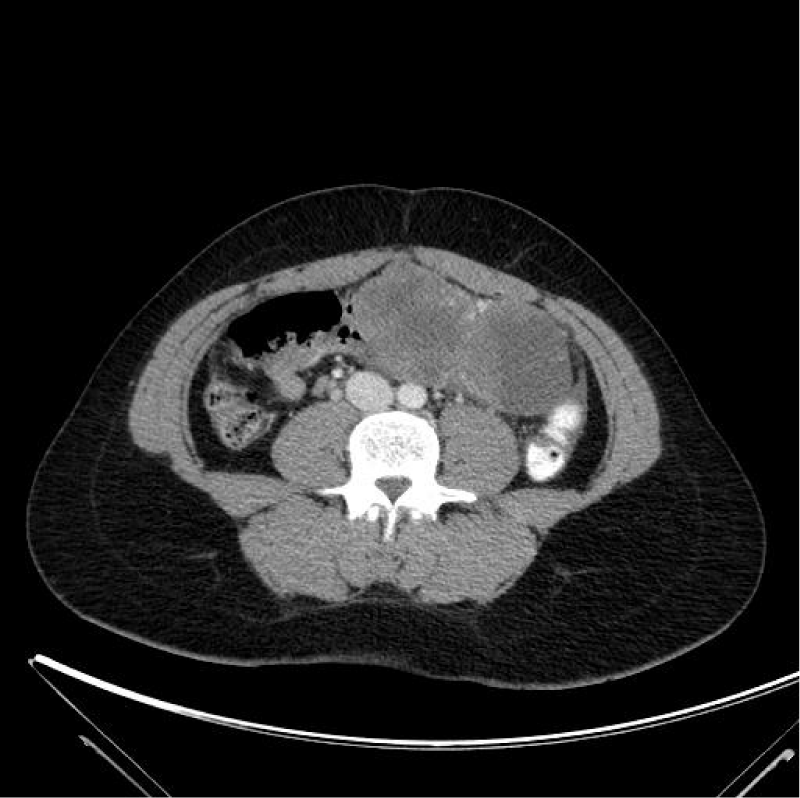
Figure 1: Abdominal CT-scan: large heterogeneous right adnexal mass. No evidence of ascites and carcinomatous.
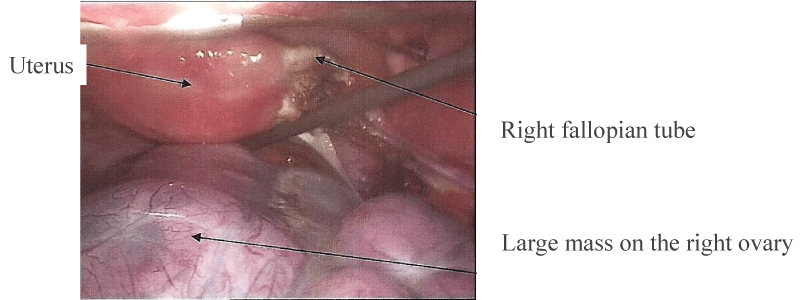
Figure 2: Abdominal CT-scan: large heterogeneous right adnexal mass. No evidence of ascites and carcinomatous.
The pathological analysis confirmed a right ovarian mass of 14 x 10 x 5 cm, weighting 380 g, with no sign of capsule rupture (Figure 3A-3B). Microscopic analysis showed the tumour was made of small cells with scanty eosinophil cytoplasm and irregular hyperchromatic nucleus, often harbouring a nucleolus. The mitotic index was high. Follicle-like cavities containing a lightly eosinophilic fluid were noticed inside the tumour (Figure 3C-3E). Immunohistochemical analysis (Table 1 for used primary antibodies) showed a prominent cytoplasmic labelling of vimentin in most cells, of synaptophysin in many cells but of chromogranin in only a few cells. Very rare cells showed cytoplasmic immunolabelling with an anti-parathormone antibody (Figure 3F-3H). A few cells showed a strong nuclear immunolabelling of p53 protein. There was no immunolabelling of cytokeratins AE1-AE3, CD99, smooth muscle actin, estrogen receptor alpha, progesterone receptor and only few cells showed immunolabelling of epithelial membrane antigen (CA 15.3). The diagnosis of small cell carcinoma of the right ovary, hypercalcemic type (SCCOHT) was proposed. Absence of immunolabelling of Brg-1 in the nucleus of neoplastic cells prompted us to look for a mutation of SMARC-A4. DNA was extracted from neoplastic cells and SMARC-A4 gene was amplified with the TruSeq Custom Amplicon kit (panel_diag_UGS_V1.1) and sequenced with Illumina MiSeq, revealing the homozygous non-sense mutation c.2866del (p. Leu956*). DNA was also extracted from peripheral blood mononuclear cells but the SMARC-A4 mutation was not found, indicating a somatic mutation in ovarian cells. No metastasis was found in the 13 lymph nodes of the right lymphadenectomy and the omentum was free of neoplastic infiltration. No cancer cell was found in the peritoneal fluid. Tumour was thus staged as FIGO IA.
| Table 1: List of used antibodies. | ||||
| Antigen | Host/Ig type | Clone | Source | Dilution |
| Bcl-2 | Mouse IgG1, k | 124 | Dako | 1/18 |
| Brg-1 | ||||
| Calretinin | Rabbit Ig | Polyclonal | ImmunoLogic | 1/100 |
| CD56 (N-CAM) | Mouse IgG1 | 1B6 | NovoCastra | 1/50 |
| Chromogranin | Mouse IgG1, k | LK2H10 | Hybritech | 1/5000 |
| Cytokeratins AE1-AE3 | Mouse IgG1, k | AE1/AE3 | Dako | 1/60 |
| Cytokeratins CAM5.2 | Mouse IgG2a | CAM 5.2 | Becton Dickinson | 1/25 |
| Cytokeratins CK22 | Mouse IgG1, k | Mixture | Biomeda | 1/400 |
| Epithelial membrane antigen | Mouse IgG2a, k | E29 | Dako | 1/200 |
| Estrogen receptor alpha | Rabbit IgG | SP1 | Thermo Scientific | 1/300 |
| Inhibin alpha | Mouse IgG2a | MCA951R1 | Serotec | 1/100 |
| p16INK4a | Mouse IgG1 | G175-405 | Becton Dickinson | 1/300 |
| p53 | Mouse IgG2b, k | DO7 | Biocare Medical | 1/1000 |
| p57KIP2 | Mouse IgG2b, k | KP3 | Thermo Scientific | 1/300 |
| Placental alkaline phosphatase | Rabbit Ig | Polyclonal | Dako | 1/500 |
| Progesterone receptor | Mouse IgG1 | 1A6 | NovoCastra | 1/50 |
| Retinoblastoma gene protein | Mouse IgG1 | 13A10 | NovoCastra | 1/250 |
| Parathyroid hormone | Rabbit Ig | Polyclonal | LabVision | 1/50 |
| Synaptophysin | Rabbit Ig | Polyclonal | Zytomed Systems | 1/100 |
| Vimentin | Mouse IgG1, k | V9 | Dako | 1/400 |
| Wilm’s Tumor-1 | Mouse IgG1, k | 6F-H2 | Dako | 1/100 |
| Becton Dickinson, Erembodegem, Belgium; Biocare Medical, Concord, CA, USA; Biomeda, Foster City, CA, USA; Dako Denmark A/S, Glostrup, Denmark; Hybritech, San Diego, CA, USA; ImmunoLogic, Duiven, The Netherlands; NovoCastra, Leica Biosystems, Newcastle upon Tyne, UK; LabVision, Newmarket, UK; Thermo Scientific, Fremont, CA, USA; Zytomed Systems, Berlin, Germany | ||||
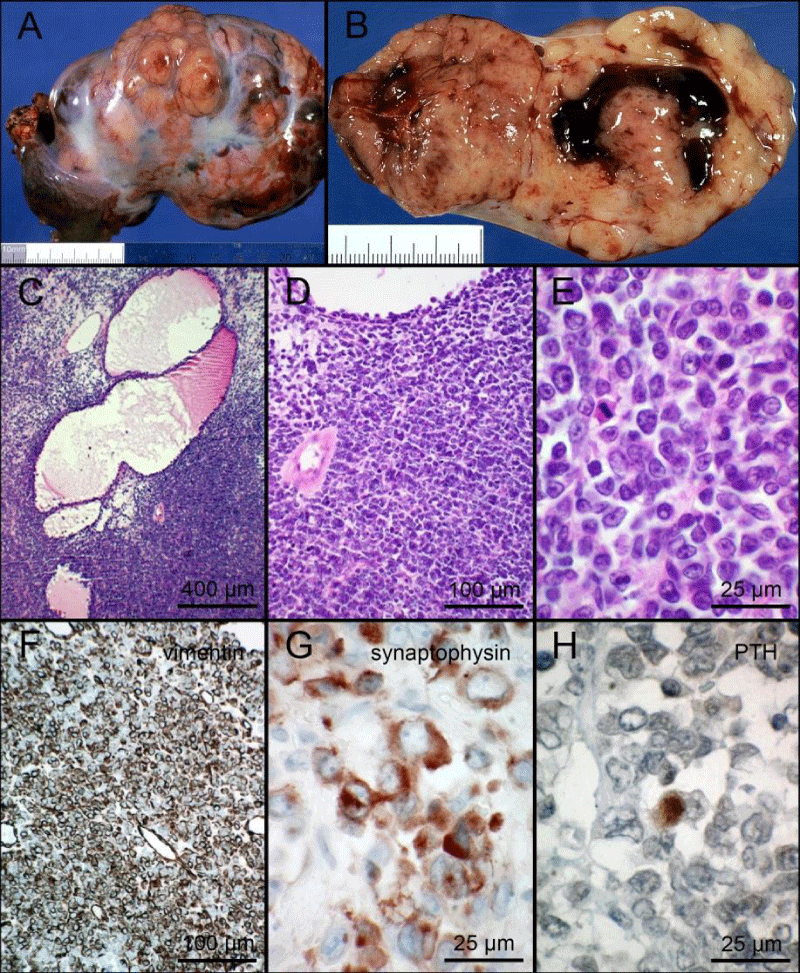
Figure 3: 3A,3B: macroscopic views of the surgical specimen (A: external view / B: section of the tumour); 3C,3D,3E: microphotographs showing small cells with scanty cytoplasm and hyperchromatic nucleus. A mitotic figure is visible at E. 3F,3G,3H: immunohistochemical labelling of vimentin (F), synaptophysin (G) and parathormone (PTH, H). Scale bars represent the indicated lengths.
Serum calcium normalized a few days later, after the patient also received hyperhydration (3 l of saline in 24 h).
Post-operatively, an 18-fluorodeoxyglucose positron emission tomography (FDG-PET) scan, was performed in order to establish the full initial extension status of this aggressive tumour. It showed no sign of metastasis or remaining disease (Figure 4).
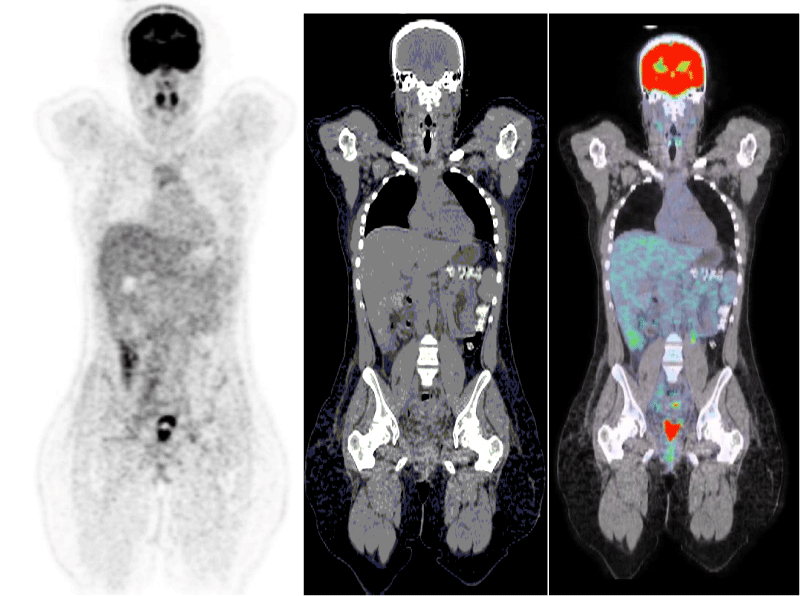
Figure 4: Pet-scan 3 days after the operation showing no sign of distant metastasis of the ovarian carcinoma.
Because of the poor prognosis, the patient then received 6 courses of chemotherapy combining cisplatin and etoposide despite the low FIGO stage. After completion of the chemotherapy, a second step surgery was performed with total hysterectomy, left salpingo-oophorectomy and left ilio-obturator and lombo-aortic lymphadenectomy. The pathological analysis showed no residual tumour and no metastasis in the lymph nodes. Upon completion of treatment, the patient was followed clinically, biologically (serum CA125 and calcium levels) as well as radiologically (thoraco-abdominal CT scan) every 3 months for 3 years and then every 6 months for up to 5 years. She is now followed clinically and biologically once a year. To date, 9 years after diagnosis, there is no sign of recurrence. Indeed, the gynecological examination, the biology and the last CT-scan were all normal. Because of her young age and to avoid premature menopausal symptoms, the patient was placed under hormonal replacement therapy in the form of a transdermal estrogen-releasing gel (Oestrogel®, Besins Healthcare) a few months after the second operation.
Discussion
We here report a case of small cell carcinoma of the ovary, hypercalcemic type (SCCOHT) with SMARC-A4 mutation, with one of the longest follow-up without recurrence of the disease found in the literature. The patient is currently alive, free of disease and in good conditions 9 years after the end of therapy. In order to conduct a complete review of SCCOHT, we searched all English and French literature on the subject (PubMed, with small cell carcinoma of the ovary of the hypercalcemic type as key words). The review is detailed in Table 2. Patient and disease characteristics are discussed below. SCCOHT is a rare tumour primarily affecting young women [1]. We found only 236 patients in the literature [Table 2], with an average age of 22.1 years. However, the disease can occur at a very young age with documented cases in a 14 month-old child [6] and an 8 year-old girl [7]. Its clinical presentation is often unspecific, with abdominal pain, nausea, weight loss and impairment of general health [1]. Some more atypical clinical manifestations have also been described such as acute pancreatitis as initial signs [8,9]. Usually, the lesion is unilateral (99% of cases), with an average size of 15 cm. half of all cases are diagnosed at stage I (two thirds at stage IA and one third at stage IC), 45% at stage III and 5% at stage II or IV [1].
| Table 2: | ||||||||
| Year of publication | Authors | Number of patients | Age | Stade FIGO | Surgery | Chemotherapy | Radiotherapy | Survival |
| 1982 | Dickersin and al, | 11 patients | average : 22 years | yes | unknown | |||
| 1994 | Young and al, | 150 patients | average : 23,9 years | 35 % de stade Ia, 15 % de stade Ib/c, 50 % de stade III et 5 % de stade II et IV | yes | unknown | ||
| 1994 | William and al, | 1 patient | 31 years | stade IC | yes | Cisplatine/Etoposide/Bléomycine | no | 5 years without reccurence |
| 1996 | Idei Y and al, | 1 patient | 46 years | stade IIIc | no | non | no | 1 month |
| 1999 | Scott and al, | 1 patient | 14 months | stade Ia | yes | Vincristine, Etoposide, cyclo, cisplatine et doxorubicine | no | 22 months without reccurence |
| 1999 | Lamovec and al, | 2 patients | 40 et 21 years | no | ||||
| 2004 | Lei Chen and al, | 1 patient | 37 years | stade IC | yes | Paclitaxel/ Carboplatine | no | 27 months without reccurence |
| 2004 | Bourgain and al, | 1 patient | 19 years | stade Ic | yes | no | no | 20 months |
| 2004 | Wynn and al, | 1 patient | 27 years | yes | Carboplatine / Placlitaxel | no | 16 months without reccurence | |
| 2005 | Harrison and al, | 17 patients | average : 34,5 years | 70% stade I et 25 % stade III and 5 % unknown | yes | yes | 60% yes | unknown |
| 2005 | Popiolek and al, | 1 patient | 35 years | stade III | yes | Cisplatine/ Etoposide | no | 22 months without reccurence |
| 2005 | Kedzia and al | 2 patients | 13 et 16 years | stade Ic et stade IIIb | yes | yes | no | < 1 year ( 2x) |
| 2006 | Chen Fan and al, | 1 patient | 26 years | stade Ia | yes | unknown | unknown | unknown |
| 2007 | Pautier and al, | 27 patients | Average : 25 years | 5 stade I,4 stade II c, 17 stade III/IV and one unknown | yes | PAVEP | yes | Average 30 months |
| 2008 | Isonishi and al | 3 patients | 24,37,25 years | IIC, IIIC, IIIc | yes | yes | no | >4 years, 4 et 9 months |
| 2008 | Cheng et al | 1 patient | 14 years | stade IV | yes | yes | no | 5months |
| 2012 | Matt McDonald et al, | 2 patients | 23 et 11 years | stade IIC / stade Ic | yes | Cisplatine/ paclitaxel | yes | 11 and 18 months |
| 2012 | Zaied and al, | 1 patient | stade Ic | yes | Etoposide/Bleomycine/Cisplatine | no | 2 months | |
| 2012 | Arvind Bakru and al, | 1 patient | stade IIIa | yes | VPCBAE | no | 6 months | |
| 2012 | Wallbillich and al, | 3 patients | 24,28,26 years | unknown | yes | yes ( VPCBAE) | no | 33, 16 and 1 months without reccurence |
| 2012 | Stephens B | 1 patient | 21 years | stade IIIC | yes | VPCBAE | yes | 5 months |
| 2012 | Woopen and al, | 4 patients | 25,29,18 and 24 years | stade Ia (2x), stadeIIb, stade IIIc | yes | PAVEP/cispl,etoposide, paclit, carbo | no | 22 months ( 3x) and 15 months for stade IIIc |
| 2012 | Otte A and al, | 1 patient | 31 years | stade Ia | yes | no | no | 13 months |
| 2012 | Bakhru and al | 1 patient | 14 years | stade IIIa | yes | VPCBAE | no | 6 months |
| 2013 | Kopp and al, | 1 patient | 5 years | stade IV | yes | cispl/doxo/etoposide/ cyclo+ graft original cells | no | 6 months |
| 2014 | Gribi and al | 2 patientes | 10,11 years | stade a | yes | vinblastin, cisplatin , bleomycine | no | 3years and 7 years without reccurence |
| 2014 | Bahri and al, | 1 patient | 34 years | stade Ia | yes | PAVEP | yes | 9 months without reccurence |
| 2015 | Callegaro-Filho and al, | 47 patients | Median age : 30 years | 34% stade I, 12,8% stade II,48,9% stade III and 4,3 % stade IV | yes ( 83,3% chemotherapy alone / 9,5% chemotherapy and radiotherapy | Cisplatin/Carboplatin/Etoposide( 38,8%) / VPCBAE ( 25,6%) | yes (9,5%) | 14 months |
| 2015 | Lavrut and al, | 1 patient | 14 years | stade IV | yes | PAVEP-CARBOPEC regimen | yes | 4 months |
| 2015 | Moens-Sosnowska and al | 2 patients | 21 years and 35 years | stade IV and IIIB | no, yes | 6 placlitaxel + carboplatin/ 3 lines of combination chemotherapy | no | 6 months/ 1 year |
| 2015 | Park and al, | 1 patient | 19 years | no specified | yes | no specified | no specified | no specified |
| 2016 | Kascak and al, | 1 patient | 24 years | no specified | yes | Cisplatin/Etoposide | no | 10 months |
| 2016 | wand and al | 1 patient | 29 years | no specified | yes | Carboplatin / Placlitaxel | no | alive |
| 2016 | Steward and al | 1 patient | 14 years | stade Ia | yes | bleomycine, etoposide and,cipslatine - BEP | no | 11 years without reccurence |
| 2017 | Witkowski | 4 patients | 28 years | stade IC, IIIb( 2x), unknow | yes | cisplatin/Etoposide | yes | alive,3 months,14 months and 3 weeks |
| 2017 | Ghazi and al, | 1 patient | 35 years | stade IIIc | no | no | no | no specified |
| 2018 | Gerday et al, | 1 patient | 23 years | Stade Ia | Yes | Cisplatin / Etoposide | no | 6 years |
In two-thirds of cases, hypercalcemia is observed but only a minority of patients is symptomatic at the time of diagnosis. Symptoms of increased calcium levels include disorders of consciousness, confusion, and sometimes polyuria and polydypsia. However, this type of non-parathyroid hypercalcemia is largely asymptomatic and will only be detected on blood analysis. The pathophysiological mechanism is not fully understood but it could result from the secretion by the tumour cells of a protein closely related to parathyroid hormone (PTH) namely parathyroid hormone-related protein (PTHrp). The protein can be detected in tumour cells by immunohistochemistry [10] and its mRNA by in situ hybridization [11]. Several studies suggested that the serum level of PTHrp and calcium could be a biological tumour marker [1,10,11]. In our case, immunostaining of PTH in very few tumour cells is consistent with this hypothesis [Figure 3H]. Normalization of serum calcium level is frequently found after surgical resection of the tumour, as in our clinical case, suggesting secretion of PTHrp by the tumour cells. Medical therapies, such as hyperhydration, can also be used before surgery to reduce hypercalcemia, with addition of diuretics like furosemide or administration of calcitonin and pamidronate in severe cases [6]. In our patient, the calcium level was high at diagnosis and normalized after surgery. Since then, her serum calcium level remained normal.
Macroscopically, the tumour is usually large, unilateral, solid or mixed, and can show signs of necrosis and areas of bleeding. Microscopically, the histology is specific, revealing small round cells with sparse cytoplasm and a hyperchromatic nucleus containing a small nucleolus. The mitotic index is very high. Fluid containing spaces can form between cells and create a follicle-like pattern in 80% of cases [1]. In some cases, large cells with an eosinophilic cytoplasm can be seen in a histological variant of this carcinoma. Large, typically pale eosinophilic intracytoplasmic hyaline globules are present in 60% of the tumours, usually associated with eccentric displacement of the nucleus. The nuclei are larger and less chromatic than in the small cells. This parameter is considered detrimental to patient survival [1].
The histological origin of this tumor is still controversial. In their initial report, Young et al. described the tumour as of epithelial origin. Further immunohistochemical studies showed that SCCOHT express several epithelial markers such as cytokeratins AE1/AE3 and epithelial membrane antigen, neuroendocrine markers such as synaptophysin and several other markers such as vimentin, calretinin, Wilm’s Tumour-1, CD10 and p53 [1,13]. More recent genetic studies showed a consistent mutation of the SMARC-A4 gene coding for the Brg-1 transcription factor. Mutation in the related SMARC-B1 gene is found in malignant rhabdoid tumours, and the terminology ovarian rhabdoid tumour has been proposed for SSCOHT [4].
Peritoneal small cell carcinoma, hypercalcemic type (SCC-HT) with healthy ovaries has been described by Popiołek et al. in 2005 [12]. They reported a case of a 35-year old patient who underwent exploratory laparotomy for a mass suggestive of uterine myoma. Peritoneal carcinomatosis was found and the patient had a debulking surgery. Histological analysis showed a SSC-HT with a large cell component. This is the only case reported in the literature of peritoneal SCC-HT with healthy ovaries [12]. Because of the small number of reported cases, there are presently no specific guidelines for the treatment and follow-up of patients with SCCOHT, but it is likely that such aggressive tumour requires equally aggressive treatment. In this context, three complementary treatments may be discussed: surgery, chemotherapy and pelvic radiation.
Surgery is crucial to obtain the diagnosis and to treat the disease. It is often performed in multiple steps as suggested by the study of Young et al. [1] and the present study. Indeed, most patients initially underwent unilateral salpingo-oophorectomy, sometimes with hysterectomy. Rarely, a cystectomy is first carried out [1]. In many cases, more radical surgery (with omentectomy, lymphadenectomy) is conducted in a second step, after histological confirmation of the disease. Because patients are generally young, conservative treatment is applied first but once the diagnosis is known, a second, more aggressive, surgical treatment is proposed because of the aggressiveness of the disease. The risk linked to preservation of the uterus and/or the other adnexa is not known, and there is presently no consensus regarding the safety of conservative surgery. In patients who have received conservative surgical treatment, no pregnancy has been reported after treatment. This may be explained by the chemotherapy and pelvic radiotherapy administered after the surgery as well as the low life expectancy after diagnosis.
Chemotherapy is given after surgery in the majority of patients. In Table 2 collecting all the cases described in the literature, a 99% rate of chemotherapy administration was observed. The treatment is often multidrug therapy, combining agents like cisplatin, doxorubicin, etoposide, vincristine, vinblastine and cyclophosphamide, given the aggressiveness of the tumour and the low survival rate of these patients. Several associations of cytotoxic drugs are reported in the literature such as VPCBAE (vinblastine, cisplatin, cyclophosphamide, bleomycin, doxorubicin and etoposide) [14,15] and PAVEP (cisplatin, doxorubicine, etoposide, cyclophophamide) [15]. Significant toxicity of these associations has been described such as myelosuppression, nausea and vomiting, and severe gastric disorders [16].
Table 2 shows that the most commonly used cytotoxic agents are platinum, taxol, etoposide and the previously described protocols (PAVEP and VPCBAE). Indeed, 90% of patients benefit from these agents (274 patients out of 299). The remaining 10 percent either did not receive chemotherapy (4 out of 299 patients), or the products were unknown, or they received other molecules.Chemotherapy treatments are also used for recurrence, mostly found in the pelvis and abdomen.
Pelvic radiotherapy can also be used as adjuvant therapy for patients with SCCOHT. Indeed, a prospective study conducted by Pautier et al. showed a trend in decreasing recurrence in the patients who had received radiotherapy [17]. This is the only prospective study conducted in SCCOHT, with 27 patients included. All patients received six courses of chemotherapy after the surgery. At the end of chemotherapy, those still alive without recurrence (18/27) were randomly distributed to one of two study arms: 10 patients received pelvic radiotherapy at high dose and 8 patients did not undergo any adjuvant radiotherapy. When radiotherapy was administered, only the pelvis was treated with doses between 40 Gy and 50 Gy [17]. Outcomes showed 3 recurrences among the 10 patients treated by radiotherapy but 5 recurrences among the 8 patients in the observational arm. This treatment may therefore be discussed but data are insufficient to systematically propose pelvic radiotherapy, especially to patients with early stage disease. In Table 2, 10% of patients received radiation therapy (always after chemotherapy).In the present case, no adjuvant radiotherapy was given and the patient did not experience recurrence. In case of recurrence, radiotherapy may be a treatment option if not previously used [18].
From our review of the literature, a number of disease-related and patient-related factors emerged that could influence the prognosis:
1. Tumour stage: This is a crucial element in the prognosis of the patient. In the study by Young et al. 14 out of 42 patients (33%) with stage 1A disease were still alive without recurrence after 1 to 13 years of follow-up. Conversely, only 2 out of 20 patients (10%) with stage 1C and 4 out of 62 patients (6.5%) with stage II, III and IV disease were alive at the end of the follow-up period.
2. Age at the time of diagnosis: Age below 30 years is considered to be unfavorable for patient survival.
3. Tumour size: An ovarian mass greater than 10 cm is a pejorative factor for patient survival.
4. Hypercalcemia: This represents another pejorative factor for the prognosis of the patient, especially if the calcium level is significantly elevated. Calcemia can be used as a tumour marker if it is initially raised.
5. Presence of large cells at pathologic analysis: This is considered to be unfavorable for the evolution of the disease.
6. Completeness of surgery: The more complete surgery, better the evolution of the disease.
7. The use of pelvic radiotherapy: As described in the prospective study of Pautier et al. this adjuvant therapy may bring an increase of recurrence-free survival.
In the present case report, although our patient had multiple negative factors related to the disease (age, tumour size, hypercalcemia, no radiotherapy), she is still alive and disease-free 9 years after treatment. Secondary SCCOHT lesions have also been described. The most frequent sites of metastasis are the liver, lungs, brain or bones [1], with lymph nodes involvement in some cases. One case of breast metastasis has also been reported [19]. All cases of SCCOHT with metastases show very poor survival of only a few months according to our review [1,19].
A genetic risk factor has been hypothesized to play a role in SCCOHT. Indeed, multiple cases of SCOOHT have been described within families, and often with first degree relatives [1,19]. In addition, it was observed that the age at which the disease occurred decreased in the next generation. This suggests a dominant autosomal transmission and a mutation in a gene (or a family of genes) could explain these different family cases. It has now been demonstrated that SMARC-A4 mutation is found in the majority of SCCOHT. In this context, a mutation of SMARCA4 may be a therapeutic target. Indeed, ponatinib has shown its effectiveness (inhibitor tyrosines kynases) [20].
Other previous theories suggested that p53, crucial in the genesis of many tumour diseases [18,21,22], may be impaired in these patients. Similarly, a Ras gene mutation, found in 27% of ovarian cancers, has also been described in SCCOHT [23-25]. Even if there is no clear evidence of genetic abnormality in most cases, the aggressiveness of the disease, the young age of occurrence of the disease and the tendency toward family recurrence are all arguments in favor of a genetic component in the origin of the disease [26]. In this context, McDonald and al. proposed that genetic counseling may be appropriate for the other family members [18].
Conclusion
SCCOHT is a rarely encountered tumour with only a few hundred cases reported in the medical literature. It is affecting mainly young women and has a very poor prognosis in the short term. We report a case of SCCOHT with a 9-year recurrence-free interval, which is exceptional for this pathology, and review all cases described in the literature.
Histologically, SCCOHT is composed of small cells with a hyperchromatic nucleus harbouring a nucleolus. A follicle-like pattern of tumour cells is found in 80% of cases. Immunohistochemistry is helpful to clarify the diagnosis. Hypercalcemia is found in 2/3 of cases, although it is rarely symptomatic. The main hypothesis is that the tumour secretes PTHrp, a substance closely related to PTH. Calcium levels usually normalize after tumour excision. In case of hypercalcemia identified at diagnosis, serum calcium level may be used as a tumour marker. A number of criteria influencing the prognosis of the disease have been highlighted. Treatment involves surgical removal of the tumour with total hysterectomy, bilateral salpingo-oophorectomy, pelvic and para-aortic lymphadenectomy and omentectomy followed in most cases by multi-drug chemotherapy. Adjuvant radiotherapy can also be discussed. Despite intensive treatment, the mortality rate of this disease is very high, with average survival of a few months after diagnosis.
Acknowledgement
The authors thank Mira Hryniuk for reviewing the English language of the manuscript.
References
- Young RH, Oliva E, Scully RE. Small cell carcinoma of the ovary, hypercalcemic Type. A clinicopathological analysis of 150 cases. Am J Surg Pathol. 1994; 18: 1102-1116. Ref.: https://tinyurl.com/y8nkjo8d
- Dickersin GR, Kline IW, Scully RE. Small cell carcinoma of the ovary with hypercalcemia: a report of eleven cases. Cancer. 1982; 49: 188-197. Ref.: https://tinyurl.com/ycylvg6s
- Scully RE. Tumors of the ovary and maldeveloped gonads. Atlas of the tumor Pathology Fascicle. Armed Forces Institute of Pathology. 1978; 316.
- Karanian-Philippe M, Velasco V, Longy M, Floquet A, Arnould L. SMARCA4 (BRG1) loss of expression is a useful marker for the diagnosis of ovarian small cell carcinoma of the hypercalcemic type (ovarian rhabdoid tumor): a comprehensive analysis of 116 rare gynecologic tumors, 9 soft tissue tumors, and 9 melanomas. Am J Surg Pathol. 2015; 39: 1197-1205. Ref.: https://tinyurl.com/yb4cxnhg
- Dykgraaf RH, de Jong D, van Veen M, Ewing-Graham PC, Helmerhorst TJ, et al. Clinical management of the ovarian small-cell carcinoma of hypercalcemic type the: a proposal for conservative surgery in an advanced stage of disease. Int J Gynecol Cancer. 2009; 19: 348-353. Ref.: https://tinyurl.com/ycjxz32t
- Florell SR, Bruggers CS, Matlak M, Young RH, Lowichik A. Ovarian small cell carcinoma of hypercalcemic type in the a 14 month old: the youngest reported case. Med Pediatr Oncol. 1999; 32: 304-307. Ref.: https://tinyurl.com/y7eape67
- Schleef J, Wagner A, Kleta R, Schaarschmidt K, Dockhorn-Dworniczak B, et al. Small-cell carcinoma of the ovary of the hypercalcemic type in an 8-year-old girl. Pediatr Surg Int. 1999; 15: 431-434. Ref.: https://tinyurl.com/y8n9deqm
- Wynn D, Everett GD, Boothby RA. Small cell carcinoma of the ovary with hypercalcemia causes severe pancreatitis and altered mental status. Gynecol Oncol. 2004; 95: 716-718. Ref.: https://tinyurl.com/y8a48vsq
- Bourgain A, Acker O, Lambaudie E, Boukerrou M, Chevalier-Place A, et al. Small cell carcinoma of the ovary of the hypercalcemic type revealed by a severe acute pancreatitis: about one case. Gynecol Obstet Fertil. 2005; 33: 35-38. Ref.: https://tinyurl.com/y7n53lyn
- Lei CH, Tri A, Abida H. Small cell carcinoma of the ovary with hypercalcemia and ectopic Parathyoid hormone Production. Arch Pathol Lab Med. 2005; 129: 531-533. Ref.: https://tinyurl.com/yd5qkr3l
- Tsunematsu T, Saito T, Iguchi H, Fukuda T, Tsukamoto N. Hypercalcemia due to parathyroid hormone-related protein produced by primary ovarian clear cell adenocarcinoma: case report. Gynecol Oncol. 2000; 76; 218-222. Ref.: https://tinyurl.com/yd4z2ep7
- Popiołek DA, Kumar AR, Mittal K. Large cell variant of small cell carcinoma, hypercalcemic type, of primary peritoneal origin. Gynecol Oncol. 2005; 96: 249-253. Ref.: https://tinyurl.com/y9kh3c6g
- Harrison ML, Hoskins P, du Bois A, Quinn M, Rustin GJ, et al. Small cell of the ovary, hypercalcemic type-Analysis of combined experience and recommendation for management. AGCIG study. Gynecol Oncol. 2006; 100: 233-238. Ref.: https://tinyurl.com/y7dt2bab
- Wallbillich JJ, Nick AM, Ramirez PT, Watkins JL, Euscher ED, et al. Vinblastine, cisplatin, cyclophosphamide, Bleomycin, Doxorubicin, and Etoposide (VPCBAE) in the management of three patients with small-cell carcinoma of the Ovary. Gynecol Oncol Case Rep. 2012; 2: 58-60. Ref.: https://tinyurl.com/ycczq357
- Estel R, Hackethal A, Kalder M, Münstedt K. Small cell carcinoma of the ovary of the hypercalcemic type: an analysis of clinical and prognostic aspects of a rare disease on the basis of cases published in the literature. Gynecologic Oncology. 2011: 284: 1277-1282. Ref.: https://tinyurl.com/ydeaj64h
- Reed WC. Small cell carcinoma of the ovary with hypercalcemia: report of a case of survival without recurrence 5 years after surgery and chemotherapy. Gynecol Oncol. 1995; 56: 452-455. Ref.: https://tinyurl.com/ybuw2f8h
- Pautier P, Ribrag V, Duvillard P, Rey A, Elghissassi I, et al. Results of a prospective dose-intensive regimen in 27 patients with small cell carcinoma of the ovary of the hypercalcemic type. Ann Oncol. 2007; 18: 1985-1989. Ref.: https://tinyurl.com/y9v2sb2x
- McDonald JM, Karabakhtsian RG, Pierce HH, Iocono JA, Desimone CP, et al. Small cell carcinoma of the ovary of the hypercalcemic type: a case report. J Pediatr Surg. 2012; 47: 588-592. Ref.: https://tinyurl.com/y9xmony4
- Cheng Z, Yin H, Du J, Yue X, Qian X, et al. Bilateral breast metastasis from small cell carcinoma of the ovary. J Clin Oncol. 2008; 26: 5129-5130. Ref.: https://tinyurl.com/y6wdza3y
- Lang JD, Hendricks WPD, Orlando KA, Yin H, Kiefer J, et al. Ponatinib shows potent antitumor activity in small carcinoma of the ovary hypercalcemic type (SCCOHT) through multi-kinase inhibition. Clin Cancer Res. 2018; 24: 1932-1943. Ref.: https://tinyurl.com/ycrtupk4
- Lamovec J, Bracko M, Cerar O. Familial occurrence of small cell carcinoma of the ovary. Arch Pathol Lab Med. 1995; 119: 523-527. Ref.: https://tinyurl.com/y6w9q9s5
- Bahri M, Lahmar R, Ben Salah H, Kallel N, Ben Amar M, et al. Small cell carcinoma of the ovary. Cancer Radiother. 2014; 18: 198-200. Ref.: https://tinyurl.com/ybo9mqg7
- Idei Y, Kitazawa S, Fujimori T, Ajiki T, Asaka K, et al. Ovarian small cell carcinoma with K-ras mutation: a case report with genetic analysis. Hum Pathol. 1996; 27: 77-79. Ref.: https://tinyurl.com/ybrlzxhl
- Witkowski L, Carrot-Zhang J, Albrecht S, Fahiminiya S, Hamel N, et al. Germline and somatic mutations characterize small cell carcinoma of the ovary, hypercalcemic type SMARCA4. Nat Genet. 2014; 46: 438-443. Ref.: https://tinyurl.com/y8zg2o5a
- Ramos P, Karnezis AN, Craig DW, Sekulic A, Russell ML, et al. Small cell carcinoma of the ovary, hypercalcemic type, displays frequent f-germline and somatic mutations in SMARCA4-inactivating. Nat Genet. 2014; 46: 427-429. Ref.: https://tinyurl.com/yc92mf8f
- Otte A, Göhring G, Steinemann D, Schlegelberger B, Groos S, et al. A tumor-derived population (SCCOHT-1) as model cellular for a small cell ovarian carcinoma of the hypercalcemic type. Int J Oncol. 2012 ; 41: 765-775. Ref.: https://tinyurl.com/y7htoter